This page serves as an index for the applications written and distributed by the Yandell, Marth, and Quinlan labs. Each item may include links to: documentation, code, and publications.
Software is listed with most recent releases first.

VARPRISM (VARiant PRIoritization SuM)
Variant Prioritization
A software package that identifies genes with a statistical excess of damaging de novo mutations among individuals with a genetic disease. VARPRISM incorporates functional variant prediction information (the VAAST CASM score) to improve the statistical power of risk gene mapping and controls for local mutation rate heterogeneity. The beta version of VARPRISM is currently available for download.
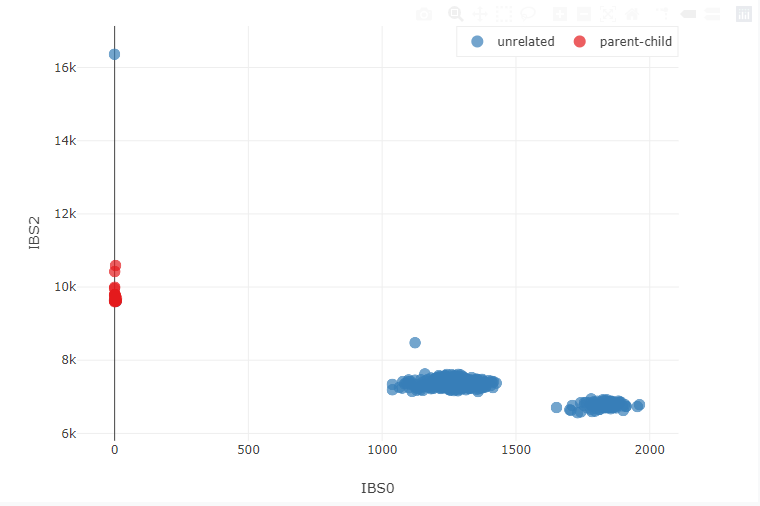
SOMALIER
Extract informative sites, evaluate relatedness, and perform quality-control on BAM, CRAM, BCF, VCF, and GVCF. somalier makes checking any number of samples for identity easy directly from the alignments.
SLIVAR
Search, and install genomic data packages. Build and check new ggd data packages. ggd provides easy access to processed genomic data. It removes the difficulties and complexities with finding and processing the data sets and annotations germane to your experiments and/or analyses. You can quickly and easily search and install data package using ggd. ggd also offers tools to easily create and contribute data packages to ggd.

GGD
Search, and install genomic data packages. Build and check new ggd data packages. ggd provides easy access to processed genomic data. It removes the difficulties and complexities with finding and processing the data sets and annotations germane to your experiments and/or analyses. You can quickly and easily search and install data package using ggd. ggd also offers tools to easily create and contribute data packages to ggd.
D4-FORMAT
The D4 Quantatative Data Format. We sought to improve on existing formats such as BigWig and compressed BED files by creating the Dense Depth Data Dump (D4) format and tool suite. The D4 format is adaptive in that it profiles a random sample of aligned sequence depth from the input BAM or CRAM file to determine an optimal encoding that minimizes file size, while also enabling fast data access. We show that D4 uses less disk space for both RNA-Seq and whole-genome sequencing and offers 3 to 440 fold speed improvements over existing formats for random access, aggregation and summarization for scalable downstream analyses that would be otherwise intractable.

SMOOVE-NF
Nextflow implementation of the smoove workflow, integrating several other tools meant to facilate variant calling and quality control of discovered variants.

SEQCOVER
seqcover is a tool for viewing and evaluating depth-of-coverage with the following aims... show a global view where it's easy to see problematic samples and genes offer an interactive gene-wise view to explore coverage characteristics of individual samples within each gene not require a server (single html page) be responsive for up to 20 samples * 200 genes and be useful for a single-sample see how we do this highlight outlier samples based on any number of (summarized) background samples

SAMPLOT
samplot is a command line tool for rapid, multi-sample structural variant visualization. samplot takes SV coordinates and bam files and produces high-quality images that highlight any alignment and depth signals that substantiate the SV.
ONCOGEMINI
OncoGEMINI is an adaptation of GEMINI intended for the improved identification of biologically and clincally relevant tumor variants from multi-sample and longitudinal tumor sequencing data. Using a GEMINI-compatible database (generated from an annotated VCF file), OncoGEMINI is able to filter tumor variants based on included genomic annotations and various allele frequency signatures.

MOSDEPTH
Fast BAM/CRAM depth calculation for WGS, exome, or targeted sequencing. mosdepth can output... per-base depth about 2x as fast samtools depth--about 25 minutes of CPU time for a 30X genome. mean per-window depth given a window size--as would be used for CNV calling. the mean per-region given a BED file of regions. the mean or median per-region cumulative coverage histogram given a window size a distribution of proportion of bases covered at or above a given threshold for each chromosome and genome-wide. quantized output that merges adjacent bases as long as they fall in the same coverage bins e.g. (10-20) threshold output to indicate how many bases in each region are covered at the given thresholds. A summary of mean depths per chromosome and within specified regions per chromosome. a d4 file (better than bigwig).

JIGV
igv.js server and automatic configuration to view bam/cram/vcf/bed. igv.js requires that the files are hosted on a server, like apache or nginx and it requires writing html and javascript. In a single binary, jigv provides a server and some default configuration, javascript, and HTML enabling a simple entrypoint... jigv --open-browser --region chr1:34566-34999 *.bam /path/to/some.cram my.vcf.gz
IDPLOT
Designed to accelerate SARS-CoV-2 research, idplot allows one to quickly compare similar sequences (*.fasta) to a reference (.fasta) with options to inspect recombination and similarity within an interactive report.

FREEBAYES-NF
A simplified version of freebayes-parallel written in Nextflow to handle job distribution on HPC resources. Intervals can be supplied by the user or created automatically to optimize compute utilization.

COVVIZ
A many-sample coverage browser. The aim of covviz is to highlight regions of significant and sustained deviation of coverage depth from the majority of samples.
DUPHOLD
Uphold your DUP and DEL calls. SV callers like lumpy look at split-reads and pair distances to find structural variants. This tool is a fast way to add depth information to those calls. This can be used as additional information for filtering variants; for example we will be skeptical of deletion calls that do not have lower than average coverage compared to regions with similar gc-content.
SMOOVE
smoove simplifies and speeds calling and genotyping SVs for short reads. It also improves specificity by removing many spurious alignment signals that are indicative of low-level noise and often contribute to spurious calls.
STRLING
STRling (pronounced like "sterling") is a method to detect large STR expansions from short-read sequencing data. It is capable of detecting novel STR expansions, that is expansions where there is no STR in the reference genome at that position (or a different repeat unit from what is in the reference). It can also detect STR expansions that are annotated in the reference genome. STRling uses kmer counting to recover mis-mapped STR reads. It then uses soft-clipped reads to precisely discover the position of the STR expansion in the reference genome.

INDEXCOV
Crazy fast genome coverage estimates! The BAM and CRAM formats provide a supplementary linear index that facilitates rapid access to sequence alignments in arbitrary genomic regions. Comparing consecutive entries in a BAM or CRAM index allows one to infer the number of alignment records per genomic region for use as an effective proxy of sequence depth in each genomic region. Based on these properties, we have developed indexcov, an efficient estimator of whole-genome sequencing coverage to rapidly identify samples with aberrant coverage profiles, reveal large scale chromosomal anomalies, recognize potential batch effects, and infer the sex of a sample.

GIGGLE
Giggle is Google for genomic features and intervals. That is, scalable, multi-file index for fast queries of genomic intervals.

VCFAnno
Variant Annotation
Annotates a VCF with any number of sorted and tabixed input BED, BAM, and VCF files in parallel. It does this by finding overlaps as it streams over the data and applying user-defined operations on the overlapping annotations.
Taxonomer
Metagenomics
Yandell Lab,Marth Lab,Eilbeck Lab
Taxonomer is an ultrafast web-tool for comprehensive metagenomics data analysis and interactive results visualization. Taxonomer is unique in providing integrated nucleotide and protein-based classification and simultaneous host mRNA transcript profiling.
RUFUS
Variant Calling
A new approach to variant detection that does not rely on mapping or whole genome assembly methods.
WHAM (WHole-genome Alignment Metrics)
Variant Calling
A structural variant (SV) caller that integrates several sources of mapping information to identify SVs. WHAM classifies SVs using a flexible and extendable machine-learning algorithm (random forest).

Genome Query Tools (GQT)
Data Management,Query Tools
A command line tool and a C API for storing and querying large-scale genotype data sets like those produced by 1000 Genomes, the Uk100K, and forthcoming datasets involving millions of genomes.
SpeedSeq
Variant Annotation,Variant Calling
An open-source genome analysis platform that accomplishes alignment, variant detection and functional annotation of a 50× human genome in 13 h on a low-cost server and alleviates a bioinformatics bottleneck that typically demands weeks of computation with extensive hands-on expert involvement.
Peddy
Pedigree Analysis
Compares familial-relationships and sexes as reported in a PED file with those inferred from a VCF.
MAKER-P
Genome Annotation
A pipeline designed to make the annotation of novel plant genomes tractable for small groups with limited bioinformatics experience and resources, and faster and more transparent for large groups with more experience and resources.
Iobio
Data Visualization
iobio uses immediate visual feedback to make understanding complex genomic datasets more intuitive, and analysis more interactive.
Poretools
A flexible toolkit for exploring datasets generated by nanopore sequencing devices from MinION for the purposes of quality control and downstream analysis.

BEDTools
Data Management
A swiss-army knife of tools for a wide-range of genomics analysis tasks. Intersect, merge, count, complement, and shuffle genomic intervals from multiple files in widely used genomic file formats such as BAM, BED, GFF, VCF.

Lumpy
Variant Calling
A probabilistic framework to integrate multiple structural variation signals such as discordant paired-end alignments and split-read alignments.
pVAAST (pedigree Variant Annotation, Analysis & Search Tool)
Pedigree Analysis
A disease-gene identification tool designed for high-throughput sequence data in pedigrees.
PHEVOR (Phenotype Driven Variant Ontological Re-ranking tool)
Phenotype Tools,Variant Prioritization
Integrates phenotype, gene function, and disease information with personal genomic data for improved power to identify disease-causing alleles.
MOSAIK
A stable, sensitive and open-source program for mapping second and third-generation sequencing reads to a reference genome.

GEMINI
Data Management,Query Tools
A powerful framework for exploring genetic variation in the context of the wealth of existing genome annotations that are available for the human genome.
GPAT ++ (Genotype Phenotype Association Toolkit)
Phenotype Tools
The application of population genomics to non-model organisms is greatly facilitated by the low cost of next generation sequencing (NGS).
ImagePlane
Data Visualization
Python based software for the automated analysis of images of the animal S. mediterranea. This software allows quantification and categorization of the animal's morphology.

VAAST 2 (Variant Annotation, Analysis & Search Tool)
Variant Prioritization
Probabilistic search tool for identifying damaged genes and their disease-causing variants in personal genome sequences.
BamTools
Variant Calling
A Bayesian genetic variant detector designed to find small polymorphisms, specifically SNPs (single-nucleotide polymorphisms), indels (insertions and deletions), MNPs (multi-nucleotide polymorphisms), and complex events (composite insertion and substitution events) smaller than the length of a short-read sequencing alignment.
RepeatRunner
Genome Annotation
A CGL-based program that integrates RepeatMasker with BLASTX to provide a comprehensive means of identifying repetitive elements.
CGL (Comparitive Genomics Library, and pronounced as “Seagull”)
Provides an informatics infrastructure for a laboratory, department, or research institute engaged in the large-scale analysis of genomes and their annotations.
Scissors
A split-read aligner that maps orphaned read mates (i.e. where one end-mate is aligned with high mapping quality, but the other mate is unmapped), as well as re-maps severely clipped reads (reads mapped with many unaligned or “clipped-off” bases).